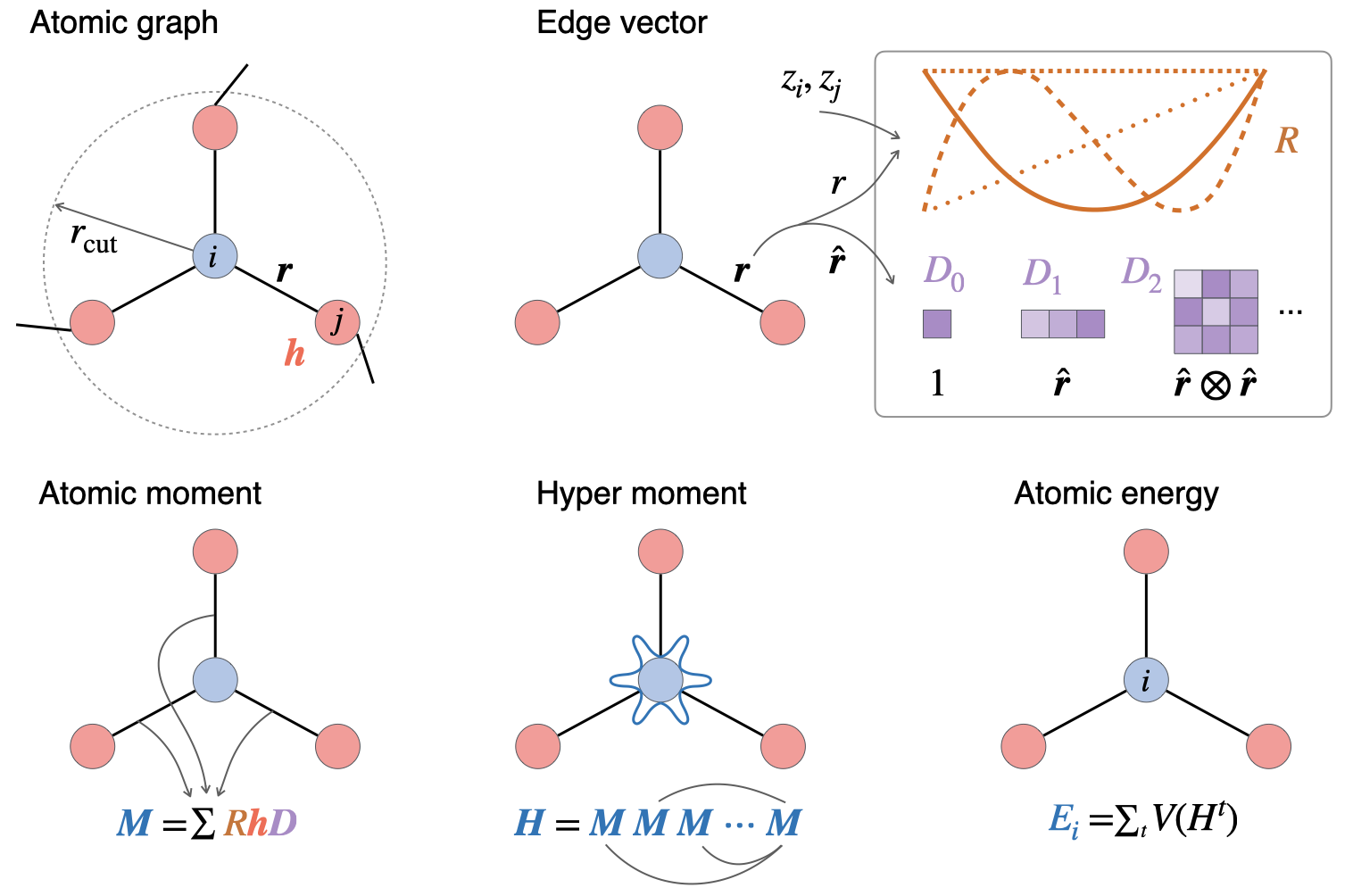
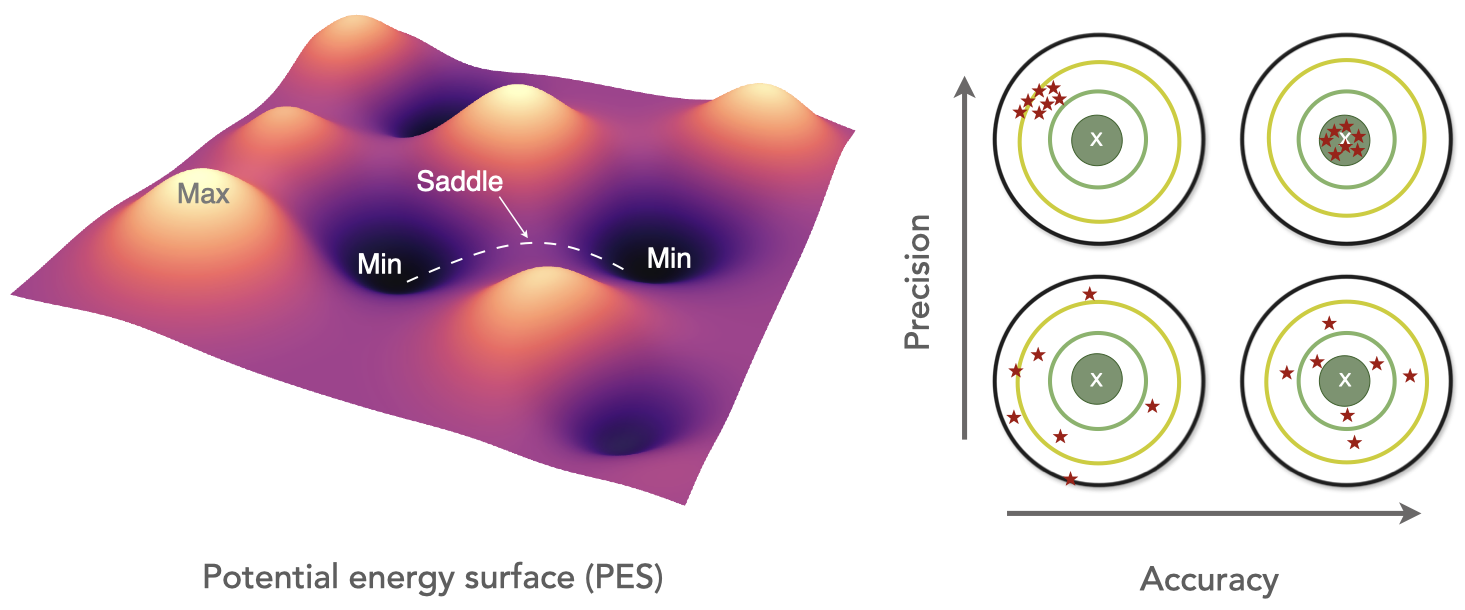
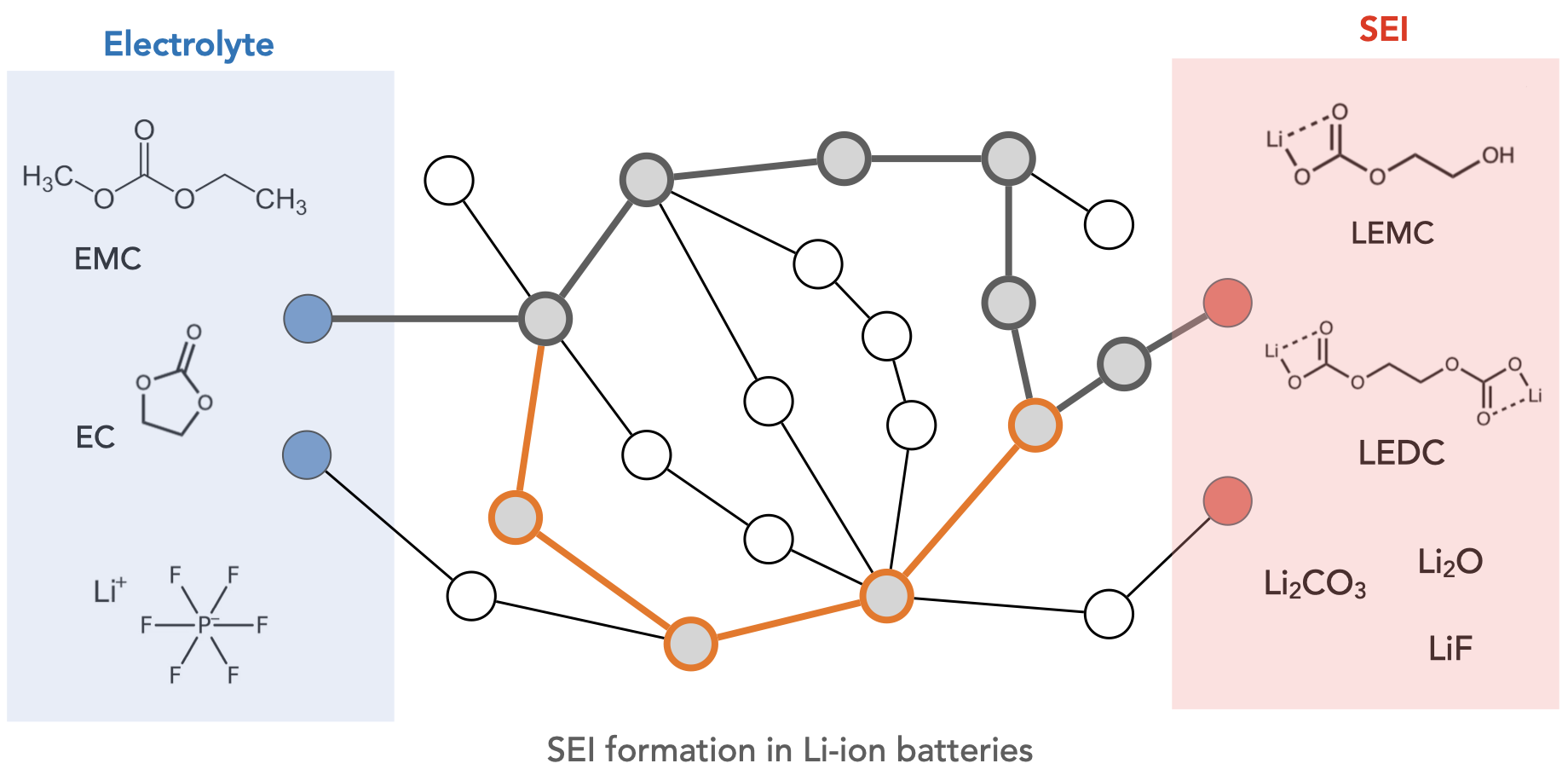
Welcome to the Wen Group at the University of Electronic Science and Technology of China!
We push the boundaries of artificial intelligence and data-driven computational methods to innovate discovery in materials and chemical sciences.
欢迎访问电子科技大学基础与前沿研究院「文明健课题组」!